Note: if you use SmCCNet in published research, please cite:
Liu, W., Vu, T., R. Konigsberg, I., Pratte, K., Zhuang Y., & Kechris, K. (2024) Smccnet 2.0: A Comprehensive Tool for Multi-omics Network Inference with Shiny Visualization. BMC Bioinformatics 25, 276.
Shi, W. J., Zhuang, Y., Russell, P. H., Hobbs, B. D., Parker, M. M., Castaldi, P. J., … & Kechris, K. (2019). Unsupervised discovery of phenotype-specific multi-omics networks. Bioinformatics, 35(21), 4336-4343.
Overview
SmCCNet is a framework designed for integrating one or multiple types of omics data with a quantitative or binary phenotype. It’s based on the concept of sparse multiple canonical analysis (SmCCA) and sparse partial least squared discriminant analysis (SPLSDA) and aims to find relationships between omics data and a specific phenotype. The framework uses LASSO (Least Absolute Shrinkage and Selection Operator) for sparsity constraints, allowing it to identify significant features within the data.
The algorithm has two modes: weighted and unweighted. In the weighted mode, it uses different scaling factors for each data type, while in the unweighted mode, all scaling factors are equal. The choice of mode affects how the data is analyzed and interpreted.
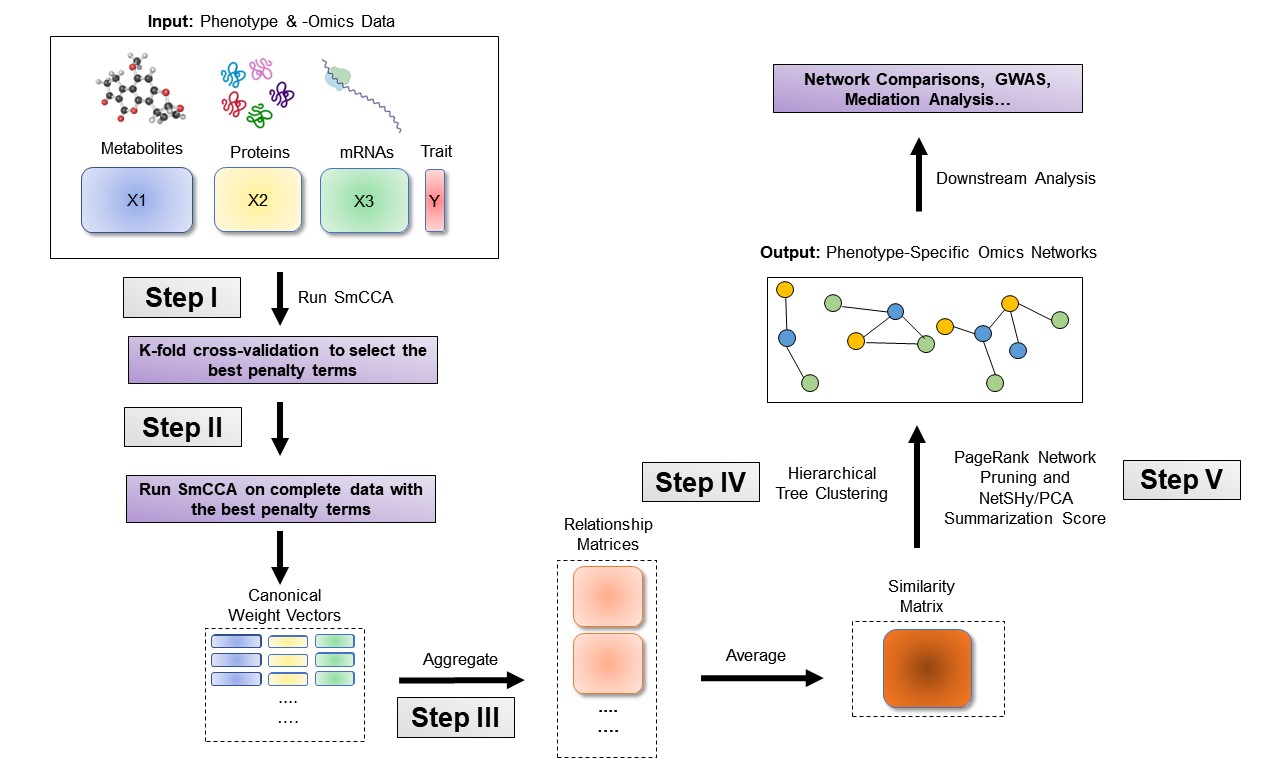
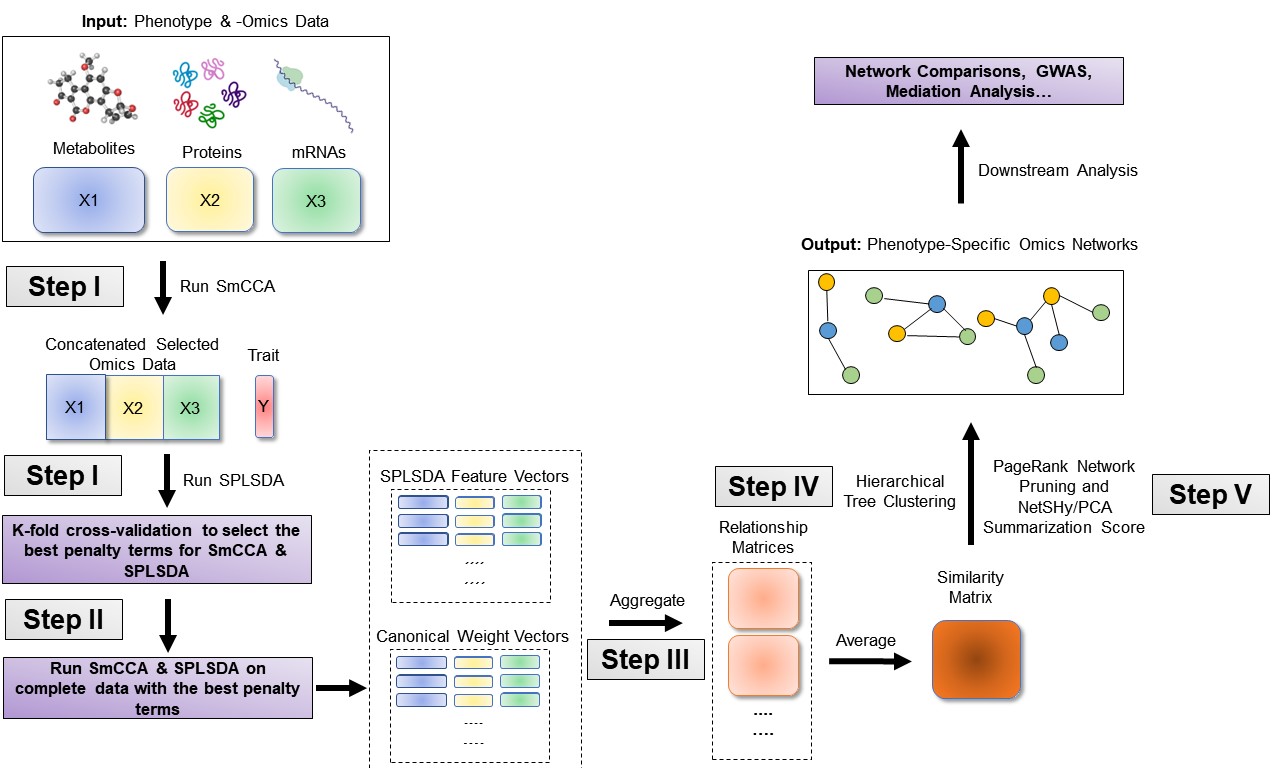
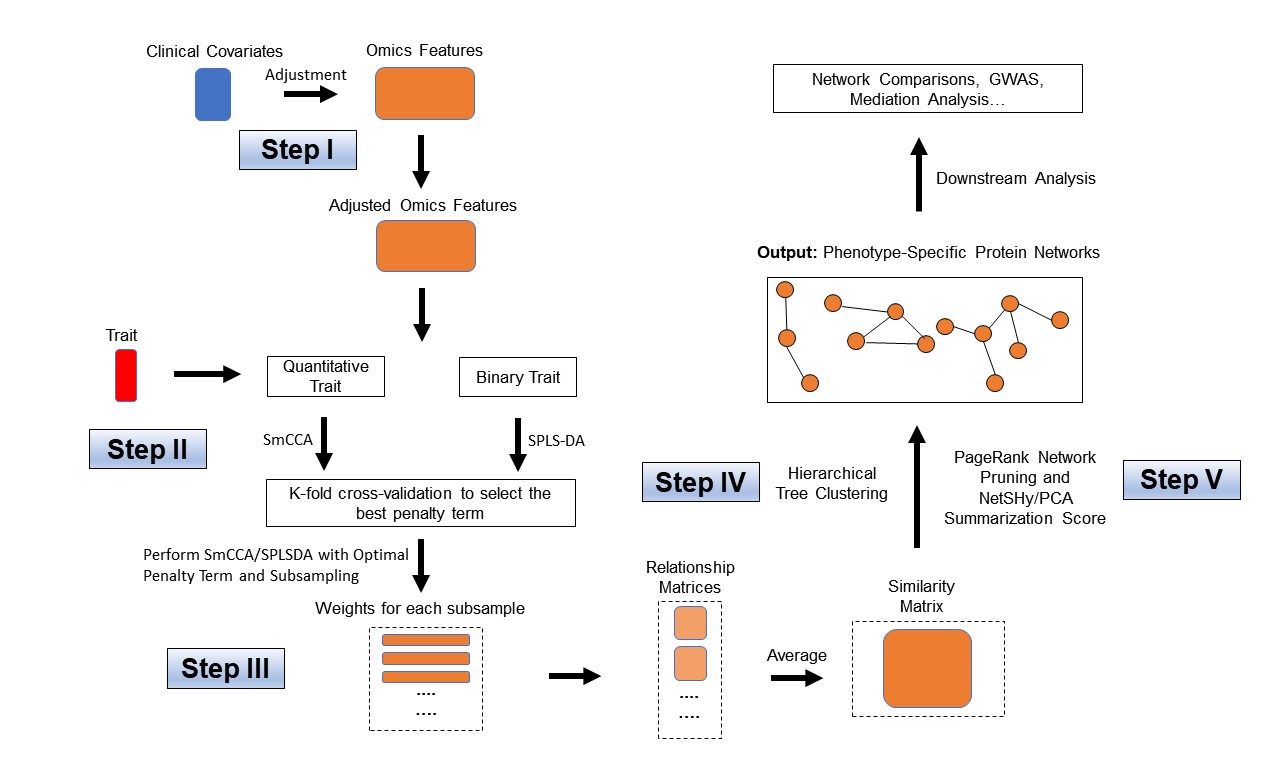
SmCCNet’s workflow consists of four main steps:
Determine Sparsity Penalties: The user selects sparsity penalties for omics feature selection, either based on study needs, prior knowledge, or through a K-fold cross-validation procedure. This step ensures the selection of features is generalizable and avoids overfitting.
Subsample and Apply SmCCA: Omics features are randomly subsampled and analyzed using SmCCA with the chosen penalties. This process is repeated multiple times to create a feature relationship matrix, which is then averaged to form a similarity matrix.
Identify Multi-Omics Networks: The similarity matrix is analyzed using hierarchical tree cutting to identify multiple subnetworks that are relevant to the phenotype.
Prune and Summarize Networks: Finally, the identified networks are pruned and summarized using a network pruning algorithm, refining the results to highlight the most significant findings.
SmCCNet Key Features
There are three major computational algorithms that are used for difrerent number of datasets and phenotype modalities:
- Sparse Multiple Canonical Correlation Analysis (SmCCA)
- Sparse Partial Least Squared Discriminant Analysis (SPLSDA)
- Sparse Canonical Correlation Analysis (SCCA)
Unlock the Power of SmCCNet with These Key Features:
- 🧬 Multi-Omics Network Inference
- With Quantitative Phenotype (SmCCA)
- With Binary Phenotype (SmCCA + SPLSDA)
- 📊 Single-Omics Network Inference
- With Quantitative Phenotype (SCCA)
- With Binary Phenotype (SPLSDA)
- 🚀 Automation Simplified
- Automated SmCCNet with a Single Line of Code
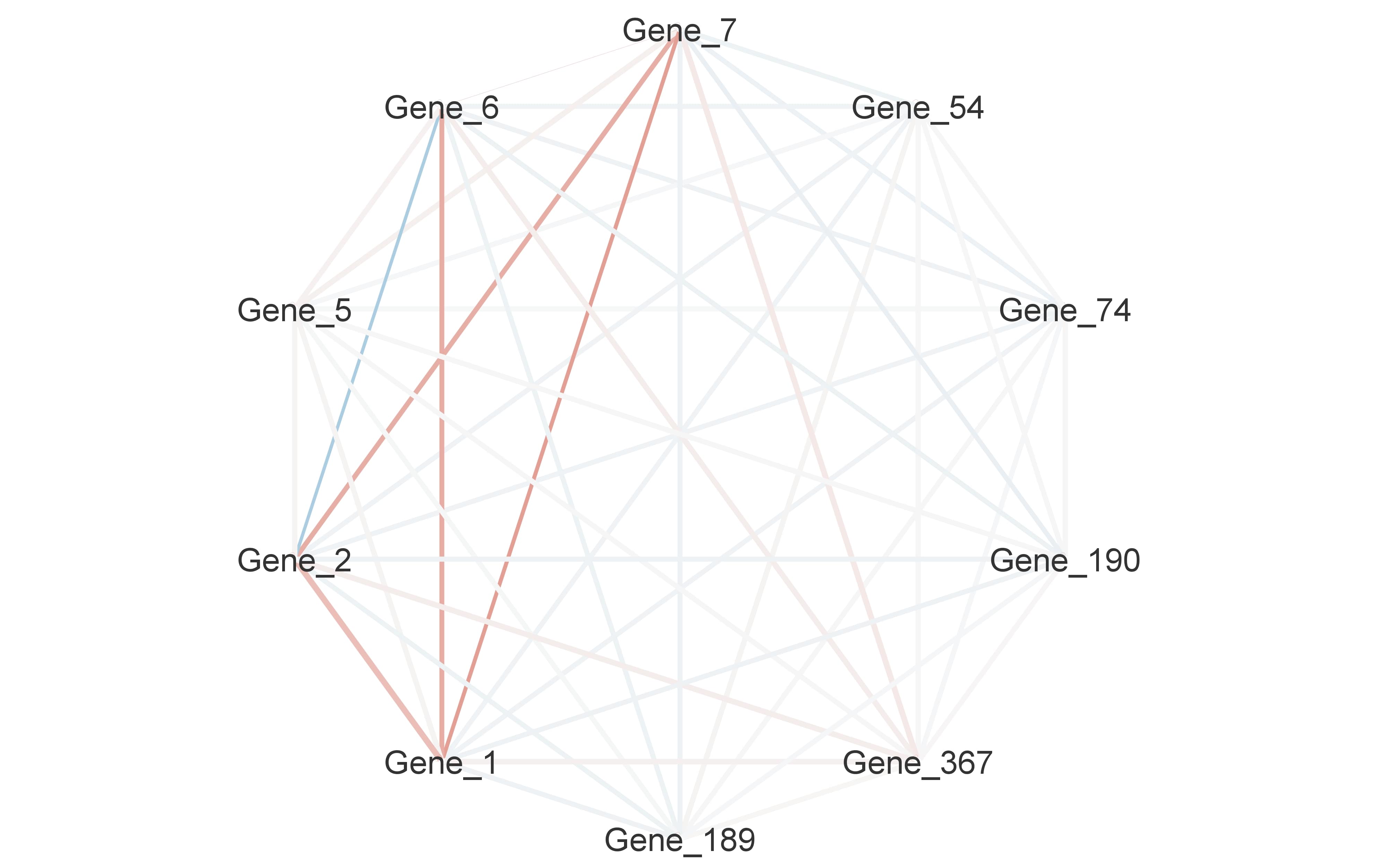
SmCCNet Network Visualization
The final network generated from SmCCNet can be visualized in two ways:
- Shiny Application simply by uplooding the final .Rdata to SmCCNet Visualization Application.
- Cytoscape Software Cytoscape through R package RCy3.

Package Functions
The older version of the SmCCNet package includes four (external) functions:
- getRobustPseudoWeights(): Compute aggregated (SmCCA) canonical weights.
- getAbar(): Calculate similarity matrix based on canonical weights.
- getMultiOmicsModules(): Perform hierarchical tree cutting on the similarity matrix and extract clades with multi-omics features.
- plotMultiOmicsNetwork(): Plot (pruned or full) multi-omics subnetworks.
In the updated package, all functions except for getAbar are retired from the package, additional functions have been added to the package to perform single-/multi-omics SmCCNet with quantitative/binary phenotype, and their use is illustrated in this vignette:
- aggregateCVSingle(): Saving cross-validation result as the cross-validation table into the working directory and provide recommendation on the penalty term selection.
- classifierEval(): Evaluate binary classifier’s performance with respect to user-selected metric (accuracy, auc score, precision, recall, f1).
- dataPreprocess(): A simple pipeline to preprocess the data before running SmCCNet (center, scale, coefficient of variation filtering and regressing out covariates).
- fastAutoSmCCNet(): Automated SmCCNet automatically identifies the project problem (single-omics vs multi-omics), and type of analysis (CCA for quantitative phenotype vs. PLS for binary phenotype) based on the input data that is provided. This method automatically preprocess data, choose scaling factors, subsampling percentage, and optimal penalty terms, then runs through the complete SmCCNet pipeline without the requirement for users to provide additional information. This function will store all the subnetwork information to a user-provided directory, as well as return all the global network and evaluation information. Refer to the automated SmCCNet vignette for more information.
- getCanWeightsMulti(): Run Sparse Multiple Canonical Correlation Analysis (SmCCA) and return canonical weight.
- getCanCorMulti(): Get canonical correlation value for SmCCA given canonical weight vectors and scaling factors.
- getRobustWeightsMulti(): SmCCNet algorithm with multi-omics data and quantitative phenotype. Calculate the canonical weights for SmCCA.
- getRobustWeightsMultiBinary(): SmCCNet algorithm with multi-omics data and binary phenotype. First, SmCCA is used to identify relationship between omics (exlude phenotype). Then, after highly connected omics features are selected in step 1, SPLSDA is used to identify relationships between these omics features and phenotype(s). The sparse PLSDA algorithm for binary outcome first compute PLS by assuming outcome is continuous, and extracts multiple latent factors, then uses latent factors to fit the logistic regression, and weights latent factor by regression parameters.
- getRobustWeightsSingle(): Compute aggregated (SmCCA) canonical weights for single omics data with quantitative phenotype.
- getRobustWeightsSingleBinary(): Compute aggregated (SmCCA) canonical weights for single omics data with binary phenotype.
- getOmicsModules(): Perform hierarchical tree cutting on the similarity matrix and extract clades with omics features.
- networkPruning(): Extract summarization scores (the first 3 NetSHy/regular prinicipal components) for specified network module with given network size. The omics features will be ranked based on PageRank algorithm, then the top omics features (where is the specified subnetwork size) will be included into the final subnetwork to generate the summarization score. For the PC score, the correlation with respect to the phenotype of interest will be calculated and stored. In addition, the correlation between individual omics features and the detected phenotype (with the Pheno argument) will also be recorded. The final subnetwork adjacency matrix will be stored into the user-specified working directory of interest.
- scalingFactorInput(): After inputing the annotation of omics data, it uses prompts to ask the user to supply the scaling factor intended for the SmCCNet algorithm to prioritize the correlation structure of interest. All scaling factor values supplied should be numeric and nonnegative.
- summarizeNetSHy(): Implement NetSHy network summarization via a hybrid approach to summarize network by considering the network topology with the Laplacian matrix.
Installation
# Install package
if (!require("devtools")) install.packages("devtools")
devtools::install_github("KechrisLab/SmCCNet")
# Load package
library(SmCCNet)Usage
We present below examples of how to execute Automated SmCCNet using a simulated dataset. In this demonstration, we simulate four datasets: two omics data and one phenotype data. We cover four cases in total, involving combinations of single or multi-omics data with either a quantitative or binary phenotype. The final case demonstrates the use of the regress-out approach for covariate adjustment. If users want to run through the pipeline step-by-step or understand more about the algorithm used, please refer to SmCCNet single or multi-omics vignettes for details.
library(SmCCNet)
set.seed(123)
data("ExampleData")
Y_binary <- ifelse(Y > quantile(Y, 0.5), 1, 0)
# single-omics with binary phenotype
result <- fastAutoSmCCNet(X = list(X1), Y = as.factor(Y_binary),
Kfold = 3,
subSampNum = 100, DataType = c('Gene'),
saving_dir = getwd(), EvalMethod = 'auc',
summarization = 'NetSHy',
CutHeight = 1 - 0.1^10, ncomp_pls = 5)
# single-omics with quantitative phenotype
result <- fastAutoSmCCNet(X = list(X1), Y = Y, Kfold = 3,
preprocess = FALSE,
subSampNum = 50, DataType = c('Gene'),
saving_dir = getwd(), summarization = 'NetSHy',
CutHeight = 1 - 0.1^10)
# multi-omics with binary phenotype
result <- fastAutoSmCCNet(X = list(X1,X2), Y = as.factor(Y_binary),
Kfold = 3, subSampNum = 50,
DataType = c('Gene', 'miRNA'),
CutHeight = 1 - 0.1^10,
saving_dir = getwd(),
EvalMethod = 'auc',
summarization = 'NetSHy',
BetweenShrinkage = 5,
ncomp_pls = 3)
# multi-omics with quantitative phenotype
result <- fastAutoSmCCNet(X = list(X1,X2), Y = Y,
K = 3, subSampNum = 50,
DataType = c('Gene', 'miRNA'),
CutHeight = 1 - 0.1^10,
saving_dir = getwd(),
summarization = 'NetSHy',
BetweenShrinkage = 5)Global network information will be stored in object ‘result’, and subnetwork information will be stored in the directory user provide. For more information about using Cytoscape to visualize the subnetworks, please refer to the multi-omics vignette section 3.1.
Getting help
If you encounter a bug, please file an issue with a reproducible example on GitHub. For questions and other discussion, please use community.rstudio.com.
This package is developed by KechrisLab, for more questions about the package, please contact Dr. Katerina Kechris or Weixuan Liu.